全球 2023-04-10
医疗器械与包装材 ISO 10993生物兼容性最新要求趋势
新版ISO 10993基本架构
ISO 10993系列标准规范了医疗器械产品的生物安全性评估程序与方法,自90年代开始一直都是医疗器械设计制造业者依循的重要安全性核心规范。自从2018年ISO组织颁布最新一版的ISO 10993-1:2018起,至今陆续在相关10993各系列标准都有重大变更,对于医疗器械行业的法规依循有了莫大的影响。新版的ISO 10993-1针对整体的医疗器械产品包含在材料本身、相关的制造程序以及工艺的残留物质以风险评量与管理的方式评价产品的生物安全性。要求医疗器械业者更加着重在材料的追溯性与资料收集的完整性。并贯彻另一主干标准ISO 14971的风险评价流程套用于整体材料的选择与使用。
目前依据ISO 10993的最新要求,执行整体生物兼容性风险评估报告前,须依据质量系统程序PDCA(Plan-Do-Check-Act)的概念进行各产品的生物兼容性评估报告。(如右图) ISO生物学测试之选择与注意事项
医疗器械制造厂或是开发者必须在设计开发前期拟定相对应的评估计划与预计的实施流程,设定该产品对应的安全性允收规格,并且依据计划内的规划进行对应的测试或是评估作业,最终将所收集的信息或是测试整合依据风险评估的原则进行完整的评估报告。在新版的ISO 10993-1强调采用ISO 14971的风险评量概念,进行一系列的生物安全因子的评估,包含生物性危害的鉴别,评断其对于人体的危害性,并且采取适当的定性与定量分析与测试结果来证明其组成含量或是溶出量的安全性。
ISO 10993-18化学表征之优先性与重点
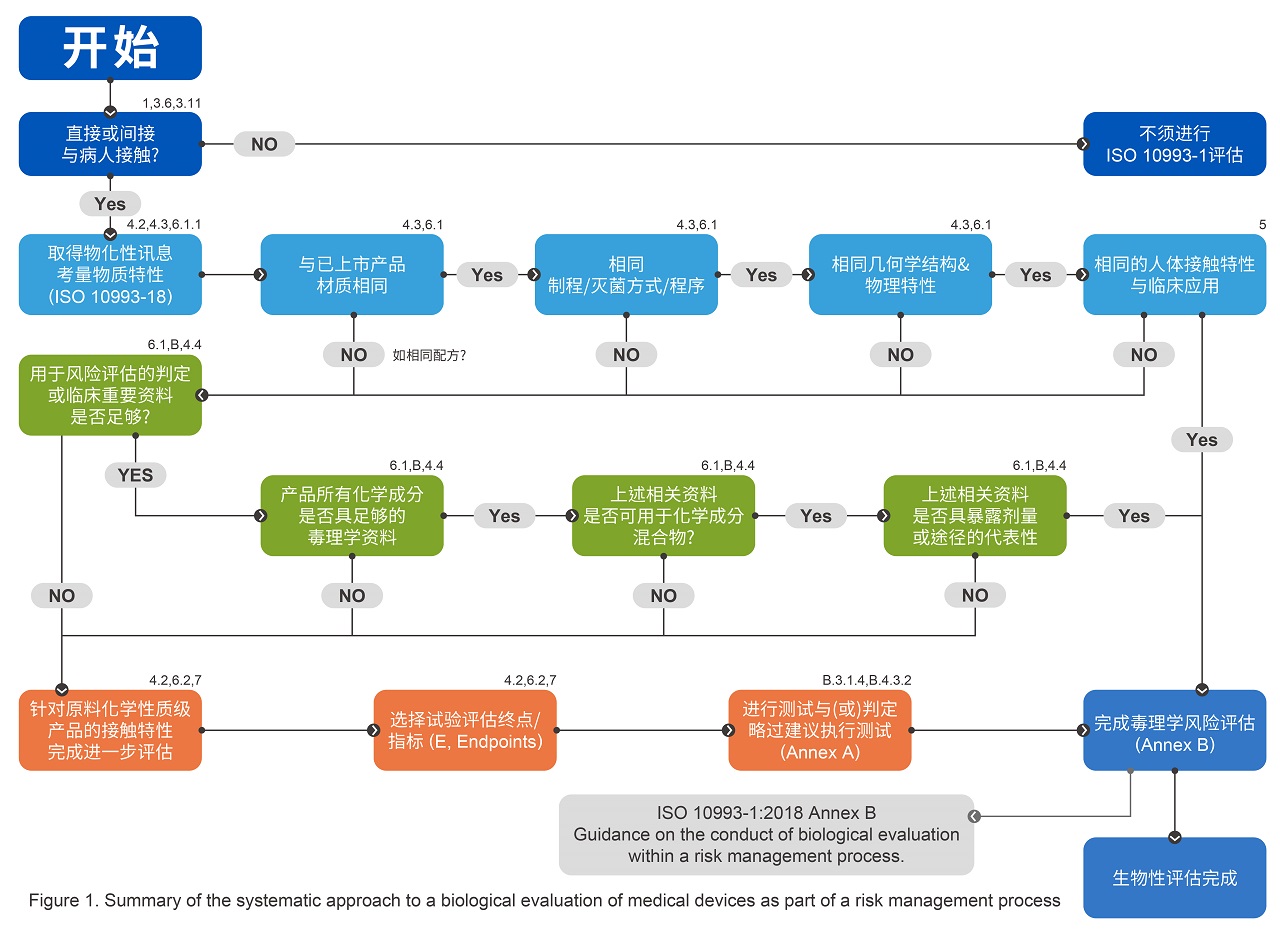
新版ISO 10993在评估流程(如下图)中开宗明义要求评估起始点要进行物理化学特性的分析与信息收集,这也是新版标准颁布以来各国主管机关采用标准的最大审查变化,此要求根据ISO 10993-18的标准描述,主要是要针对产品的「材料组成物」、「制造程序中的残留物」还有「可能的萃取物(Extractable)/溶出物(Leachable) 」收集相关的信息,其中又特别针对所谓的萃取/溶出物质需要采用毒理评估方式对其鉴别风险危害与否。
依据ISO 10993-18的规范建议,会针对无机溶出物、非挥发有机物与挥发性有机物进行定性或定量分析,目前坊间多采用高阶的化学分析质谱设备来进行鉴别,例如感应耦合电浆质谱仪(ICP-MS)、液相层析质谱仪(LC-MS)、气相层析质谱仪(GC-MS)甚至更高阶的分析设备。目的都是为了找出产品中可能的危害风险物质,并以ISO 10993-17的毒理评估流程设定每一个检出化合物可接受的允许限量,将分析的数据比对限量允收的要求进行安全限值的计算(margin of safety),并且需要进行一连串的比对已上市产品的比较,包含材质、加工制程、几何构形及物理性质与临床预期用途对应的有效性分析。
创造双赢,大于产品价值的服务价值
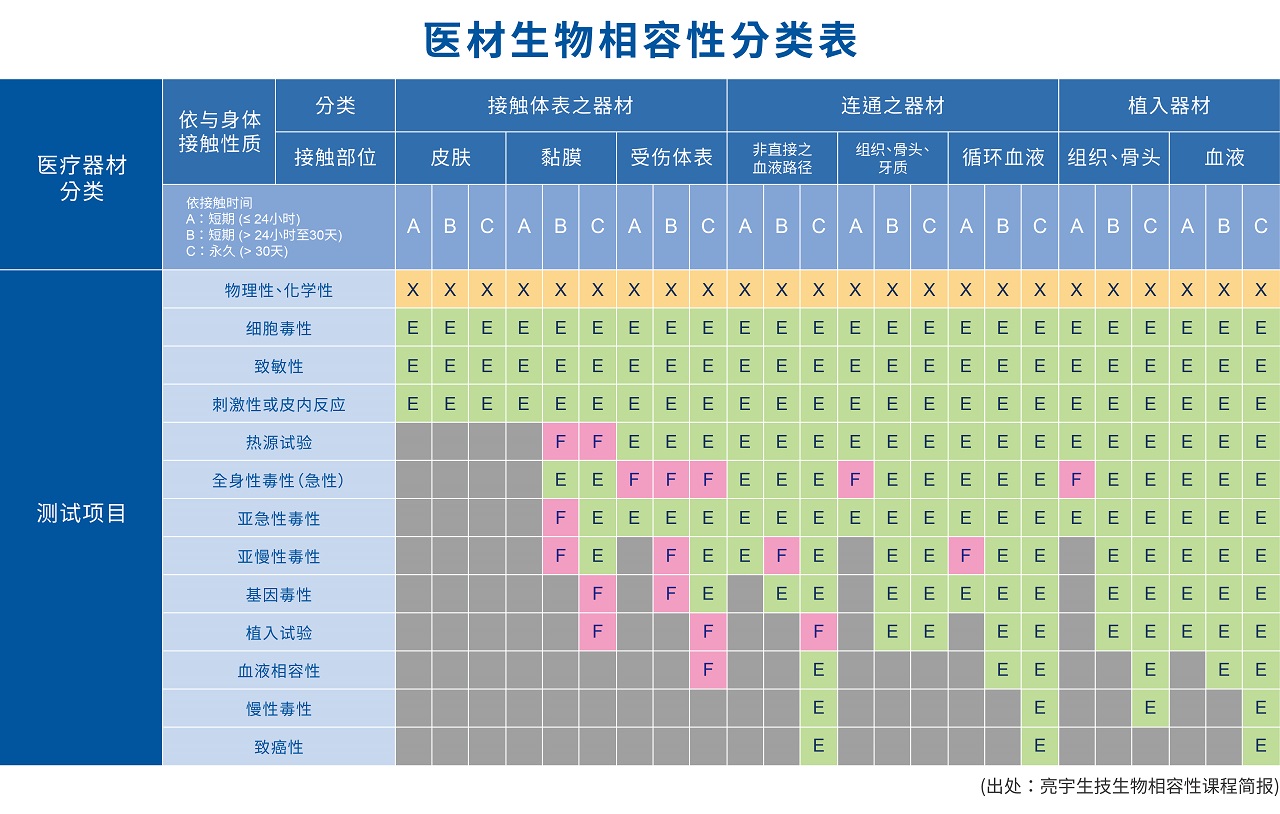
创造双赢,大于产品价值的服务价值在旧版的ISO 10993中,常以ISO 10993中表A-1的各项生物学测试当作测试的规划依据,而在新版的要求中,各项生物学的测试(表一)转变成风险评估的终点指标(end point),不再以强制要求生物测试为目的,改以要求各项生物学指标做为评估终点,医疗器械业者可以透过实质比对,既有毒理安全资料或是文献来佐证安全性的各项要求, 若无可依循的数据下,才以最终设计生物学测试的手段来满足不同产品的不同需求。 表格着重以下重点:
- 产品选用材料的物理性及化学性资料必须先收集及进行风险评估
- 新版标准的测试项目增加了热原性(Material Mediated Pyrogenicity)、慢毒(Chronic toxicity)、致癌性(Carcinogenicity) 、生殖毒性及可降解性(新材料需评估)
- “X”表示生物学风险评估的前置信息,“E”表示要在风险评估中评估的终点
- “F”则为美国FDA 的特定要求
各项试验中需要优先注意的是样品的选择与代表性,ISO 10993-12针对测试样品有规范几种样品制备的条件与要求,测试样品来源可以来自于「最终成品(无菌)」、「多种规格下代表性样品」以及「经由同样制程产出之仿真代表样品」。并且经由指定的萃取条件进行试验物质的准备。包括依据不同的萃取条件(温度与时间),还有与萃取溶液的比例)进行测试样品的制备。这部分也要依据产品材质的特性做评估与选择。
医疗包材生物兼容性之要求
在新版ISO 10993-1的标准也有提到须将与产品接触的包装材料列入评估的一环,主要是因为产品会长时间与包装材料接触,需考量包装材料是否会产生溶出物质或是迁移性的物质移转到医疗器材上,因此对于包装材料的化学分析有进行评估的必要性。
另外也须针对会直接接触医疗器械的包装材料要进行生物安全性的评估,相关资讯可参考ASTM F2475-20之准则; 此外,若是医疗器械用于药品包装上则需要依循USP<87>Biological Reactivity tests, in vitro , USP<88> Biological reactivity tests, in vivo, 进行Class I~IV因应侵入性或是植入性产品的材料安全性测试,用此以降低整体的产品风险。 另外包装材料的洁净度也跟产品的洁净度息息相关,所以在微粒残留上也须要多考量。
总结
医疗器械的风险无所不在,伴随着本次10993-1的改版对于医疗器械风险管理的要求提高,各级医疗器械生产商更需要重新在产品生命周期(Product life span)检视生产各阶段过程中可能残余的风险物质。而对于产品委托具有GLP第三方资质的实验室和测试过程中的档案保存也更趋重要,如果没有在上市前收集产品材料信息确认其安全性和降低风险,到临床医院端引起病患不良反应的后果将难以想象,这对于生产厂家的品牌声誉及信心度更会造成剧烈的影响。藉由本次10993-1改版将可使医疗器械的风险评估架构更趋完整,也符合近年来各国对于医疗器械监管的力道趋于严谨,在近期新版的欧盟MDR法规与美国US FDA对于生物兼容性的要求我们也能同步看到。随着后疫情时代健康产业的蓬勃发展,更多新型态新用途高风险的器械设计或生医新材料的开发将透过合乎法规的态度落实在临床使用的安全性上。